Hipermovilidad Articular médicos
Que significa la hipermovilidad articular
La hipermoviliad articular sin síntomas, es una buena condición, ya que permite a las personas funcionar mejor en los ejercicios y vida diaria.
La hipermovilidad articular con síntomas, pasa a ser una enfermedad, el Síndrome de Ehlers-Danlos Hipermovilidad, también llamado síndrome de Hiperlaxitud Articular o SHA.
La hipermovilidad articular indica la presencia de una alteración genética de la matriz colágena, la que forma parte de la mayoría de los tejidos. Estos se pueden distender dando esguinces, subluxaciones, dolores articulares; se pueden dilatar produciendo várices, aneurismas, disautonomía, quistes; se pueden romper, produciendo hernias; se pueden gastar, dando osteoartritis (Artrosis) y osteoporosis.
Las formas más comunes de Alteraciones Hereditarias de la Fibra Colágena son:
A.- Síndrome de Ehlers- Danlos (SED).
B.- Síndrome de Marfán (SMF).
C.- Osteogénesis Imperfecta (OI)

Que son las Alteraciones Hereditarias de la Fibra colágena
Las AHFC son enfermedades que pueden afectar múltiples órganos y que se deben a una alteración del colágeno causado por una mutación genética, que puede ser hereditaria o debida a una mutación “de novo”. La alteración del Colágeno 1 (COL1A o 2) da OI. Alteración del Colágeno II produce alteraciones de los cartílagos y displasias. La alteración del Colágeno III puede dar SEDH o EDSV (COL3A1 o 2). Desórdenes del Colágeno V (COL5A1) produce SED I-II. La alteración de la Fibrillina o Elastina puede producir SMF.
A.- Síndrome de Ehlers-Danlos
ANTIGUOS CRITERIOS DIAGNOSTICOS DEL EHLERS-DANLOS
I.- Síndrome de Ehlers-Danlos. Consistia de 10 tipos (obsoleto).
II.- Síndrome de Ehlers-Danlos. Hay 6 tipos:
"CRITERIO DIAGNOSTICO DE LOS SINDROMES DE EHLERS-DANLOS
SEGUN LA CLASIFICACIÓN DE VILLEFRANCHE 1997"
2. SED Hipermovible o SED tipo III.
4. SED Oculo-escoliótico o SED tipo VI.
1. Síndrome de Ehlers-Danlos Clásico o SED tipo I-II.
Definición. El SED Clásico es uno de los tipos de SED muy similar al SED-III, pero caracterizado por extrema hiperlaxitud y por presentar frecuentes sub-luxaciones articulares y mala cicatrización de las heridas. Se debe a una alteración del Colágeno tipo V (por mutación del gen COL5A1). Tiene herencia Autosómica dominante. Es menos frecuente que el SED-III.
Excelente artículo de los doctores Fransiska Malfait, Richard Wenstrup y Anne De Paepe.
Fotos del SED Clásico. Fotos del Dr. Bravo
2.Síndrome de Ehlers-Danlos Hipermovible o SED tipo III. También llamado Síndrome de Hipermovilidad Articular (SHA).
- Generalidades sobre Hiperlaxitud articular. Power point del artículo publicado en Arthritis & Rheumatism, 2006.
-
Tríptico - Síndrome de Ehlers-Danlos tipo III - 2018 (información en lenguaje para pacientes)
Síndrome de Hiperlaxitud Articular y Síndrome de Ehlers-Danlos Vascular (Resumen en español)
- Artículo revista de Reumatología, Mayo 2010. Epidemiología y manifestaciones clínicas.
- SED con especial énfasis en el SHA. Revista Médica de Chile.
- !!! Teleconferencia con la Asociación Española de Afectados con Hiperlaxitud (ASEDH). Barcelona. España.
Taller de cómo examinar al enfermo Hiperlaxo.
Como sospechar y diagnosticar las enfermedades de la fibra colágena: Síndrome de Ehlers-Danlos,
Síndrome de Marfán y Osteogénesis Imperfecta
Epidemiología y manifestaciones clínicas del SHA
- Facie Típica en el SHA
- Facie Típica en el SHA (II)
- Escleras Celestes
- Alteraciones de las orejas en el SHA
- Lengua movible en el SHA
- Costillas prominentes en el SHA
- Signo de la “Mano afirmando la cabeza” en el SHA
- Signo de la “Mano en forma de ave volando” en el SHA
- Habito Marfanoide en el SHA, vs. Síndrome de Marfán
- Hábito Marfanoide en el SHA
- Signos del “Pulgar horizontal” y del “Pulgar en cuello de Pato” en el SHA
- Signo de la Piel oscura en la zona de extensión de las articulaciones en el SHA: “codos oscuros”, “dedos oscuros” y “rodillas oscuras”.
- Alteraciones de la piel en el SHA
- Piel laxa en el SHA
- Lunares lenticulares en el SHA
- Venas prominentes en el SHA
- Signo de “Hombros cuadrados” en el SHA
- Signo de “Mano del escribiente laxo”
- Signo de “Huella de pata de elefante”
-
Charla para Pediatras.
-
Consecuencias físicas y psicológicas de la Pandemia y el Teletrabajo
-
Qué necesita saber el Médico General, el Internista y el Reumatólogo sobre el Ehlers-Danlos
Síndrome de Hiperlaxitud Articular (dictado en Curso del dolor),
Fotos características del SHA (SED-III):
Ayuda Visual para sospechar el SED-III, Poster presentado en Paris, Francia, Marzo 2016
Charla En Simposio Internacional Síndrome de Ehlers-Danlos e Hiperlaxitud, Murcia, España, 2015
Sugerencias para el tratamiento del SEDh (reducido)
Ejercicios para elongación de trabajadores de escritorio.
Artículos Recientes:
3.Síndrome de Ehlers-Danlos Vascular (SEDV) o SED tipo IV.
Definición. El SEDV es poco frecuente. Es más grave, por lo que es necesario hacer el diagnóstico precozmente, ya que se puede asociar a problemas cardiovasculares e incluso ruptura de órganos, como el pulmón, colon y útero grávido. Se caracteriza por tener hematomas fáciles y frecuentes, a veces sin causa aparente. Además destacan el prolapso de la válvula Mitral y condiciones más graves como aneurismas cerebrales y rupturas arteriales. En la mayoría de los casos de SEDV el diagnóstico se hace después de una ruptura arterial. El conocer el diagnóstico de SEDV antes de la complicación puede salvar la vida del paciente. Estos enfermos por lo general sólo tienen hiperlaxitud de las pequeñas articulaciones de las manos. También pueden tener Disautonomia, lo que les da una mala calidad de vida. A veces tienen la facie típica del SED Vascular: cara triangular, ojos hundidos, labio superior fino y falta de tejido adiposo de la cara. También pueden tener recesión de las encías. Para el diagnóstico, es importante la existencia de una historia familiar de complicaciones arteriales o de ruptura de órganos e incluso de muerte súbita de algún familiar menor de 30 años, sin causa aparente.
- Síndrome de Ehlers-Danlos Vascular. Articulo del Dr. Bravo en revista Chilena de Reumatología 2008
- Fotos del SED Vascular. Facies típica del SEDV.
- Criterio para diagnóstico de EDS Vascular. Criterio de Villefranche.
- Como confirmar el diagnóstico de SEDV.
- Precauciones y posibles complicaciones del SEDV. Artículo revista de cirujanos de Chile.
- Precauciones y posibles complicaciones del SEDV.
4.Ehlers-Danlos Oculo-escoliótico o SED tipo VI.
Definición. EL SED Cifoescoliótico u Oculo-Escoliótico. Antiguamente llamado SED tipo VI. Es poco frecuente y es el único SED con herencia Autosómica recesiva y se caracteriza porque el recién nacido puede tener marcada hipotonía muscular y escoliosis, o desarrollarla en el primer año de vida, asociada a problemas oculares. Tiene Osteoporosis en el 100% de los casos.
5. Síndrome de Ehlers-Danlos Artrocalasia.
Definición. SED tipo VII a
Es muy poco frecuente, con alrededor de 30 casos reportados. Afecta al tipo I del colágeno. El tipo artrocalasia se caracteriza por articulaciones muy flojas y dislocaciones con participación de ambas caderas. Herencia Autosómica dominante.
6. Síndrome de Ehlers-Danlos Dermatosparaxis
Definición. SED tipo VII b,c
También muy raro, con cerca de 10 casos reportados. El tipo dermatosparaxis se caracteriza por una piel extremadamente frágil y flácida. Herencia Autosómica recesiva.
NUEVO CRITERIO DIAGNOSTICO DEL EHLERS-DANLOS
Clasificación Internacional de los Ehlers-Danlos. 13 tipos:
(Este nuevo criterio esta diseñado para genetistas, por
su complejidad, y no es apropiado para los médicos en
general)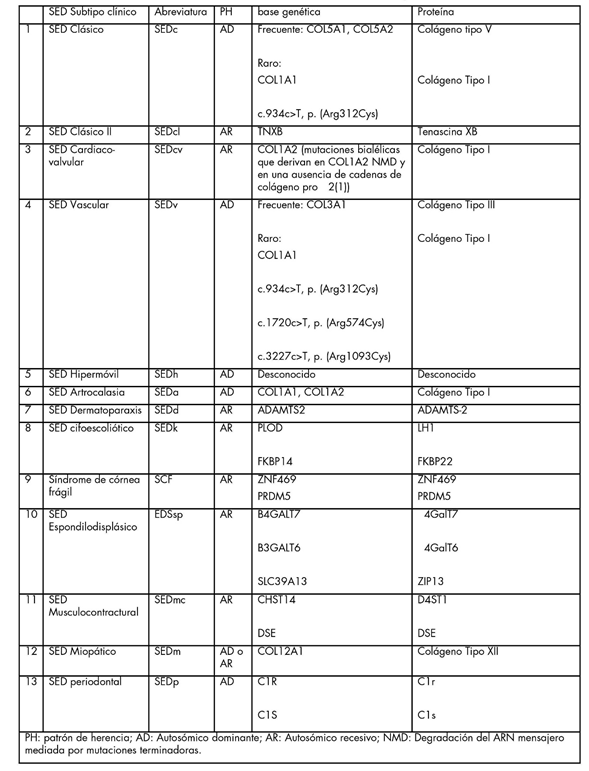
Nota: Para mayores detalles ver link del documento completo (39 páginas)
Clasificación Internacional de los Ehlers-Danlos 2017
B.- Síndrome de Marfán
El Síndrome de Marfán (SMF). Es una enfermedad hereditaria de la fibra colágena, potencialmente grave en el que algunas complicaciones importantes se pueden prevenir. Se debe a una alteración genética de la Fibrillina y Elastina. El gen alterado esta en el Cromosoma 15, Locus 21.
Es poco frecuente, uno en 12.000 personas. Tiene herencia Autosómica Dominante. En general son personas extremadamente altas y delgadas (hábito marfanoide), con la envergadura superior a la altura en 8 cm. , aracnodactilia, e hiperlaxitud articular. Puede afectar la arteria aorta, tener neumotórax espontáneo, ectopia lentis, miopía y desprendimiento de la retina. El diagnóstico se hace si existen las características típicas del aparato locomotor, más las alteraciones oculares descritas o compromiso cardiovascular. Es importante saber diferenciar los Marfanoides (SHA) de los Marfanes, ya que éstos tienen complicaciones que pueden ser mortales. La arteria aorta suele comenzar a dilatarse en la adolescencia y si no se diagnostica y trata, puede llegar en unos 10 años a la ruptura arterial y muerte por hemorragia.
Criterio diagnóstico extenso del SMF.
C.- Osteogénesis Imperfecta (OI)
Es otra enfermedad hereditaria de la fibra colágena debida a una alteración del Colágeno tipo I (Col1 o Col 2). Tienen escleras azules o celestes, pero a veces normales (blancas).Por lo general tiene herencia Autosómica Dominante y se caracteriza por tener osteoporosis severa y gran tendencia a las fracturas. Es muy poco frecuente en el adulto, uno en 100.000 personas. Se suele ver en recién nacidos y niños que se fracturan con facilidad y en forma recurrente. Afecta a 1 en 20.000 nacidos vivos. Las fracturas pueden ocurrir incluso durante la vida intrauterina. Existen 5 tipos como se puede apreciar en la Clasificación de Sillence.